1. The challenge of distinguishing physiological interfaces from crystal packing contacts
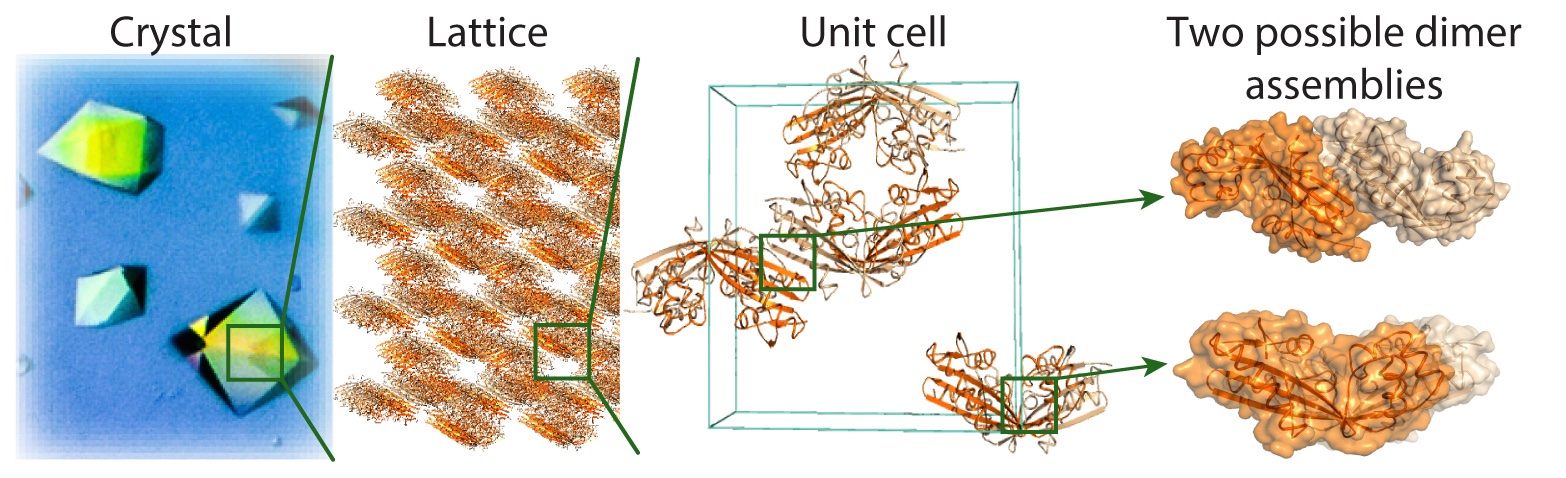
Much of our knowledge on protein quaternary structure has been derived from X-ray crystallography, from which the spatial arrangement of protein chains can be revealed (Perutz et al. 1960). Indeed, to date, over 150,000 crystallographic structures of proteins have been deposited in the Protein Data Bank (PDB) (Berman, 2000; Velankar et al., 2016). However, one caveat, of X-ray crystallography is that it requires protein molecules to be arranged in a regular array to form a crystal lattice. In this lattice, some protein-protein contacts may be part of a protein’s quaternary structure, whereas others only result from the crystal formation and are called crystal contacts. The figure below illustrates this concept: in the protein's crystal two dimer assemblies are observed in the lattice and identifying which one of the two is physiological in challenging.
2. Inferring physiological relevance from quaternary structure conservation
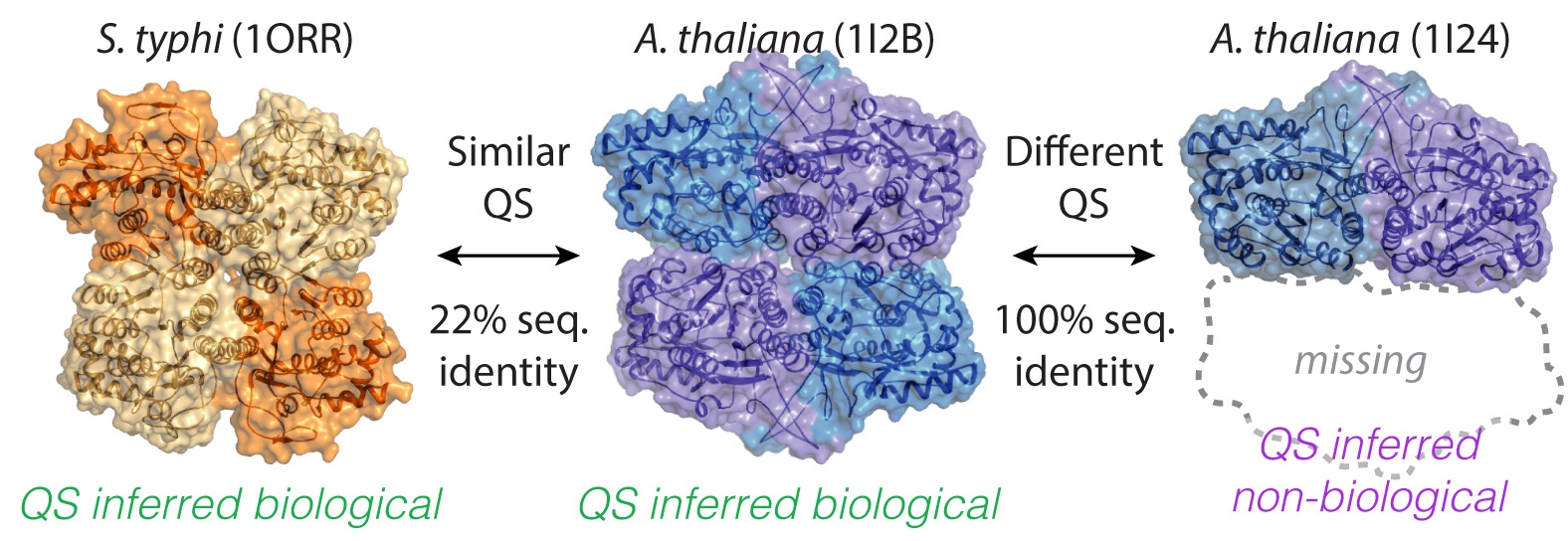
In the figure below, we illustrate how quaternary structure conservation across homologs points to physiologically relevant interfaces. Tyvelose epimerase is a tetrameric enzyme in Salmonella typhi (PDB code 1ORR). A similar tetramer is found in Arabidopsis thaliana (PDB code 1I2B, r.m.s. deviation = 3.55 Ĺ), although the sequences of these two tetramers share only 22% identity. Such conservation suggests that both tetramers are biologically relevant. This information enables subsequent correction of entries showing identical sequence but different QS (e.g., PDB code 1I24).
3. The process of QSalign
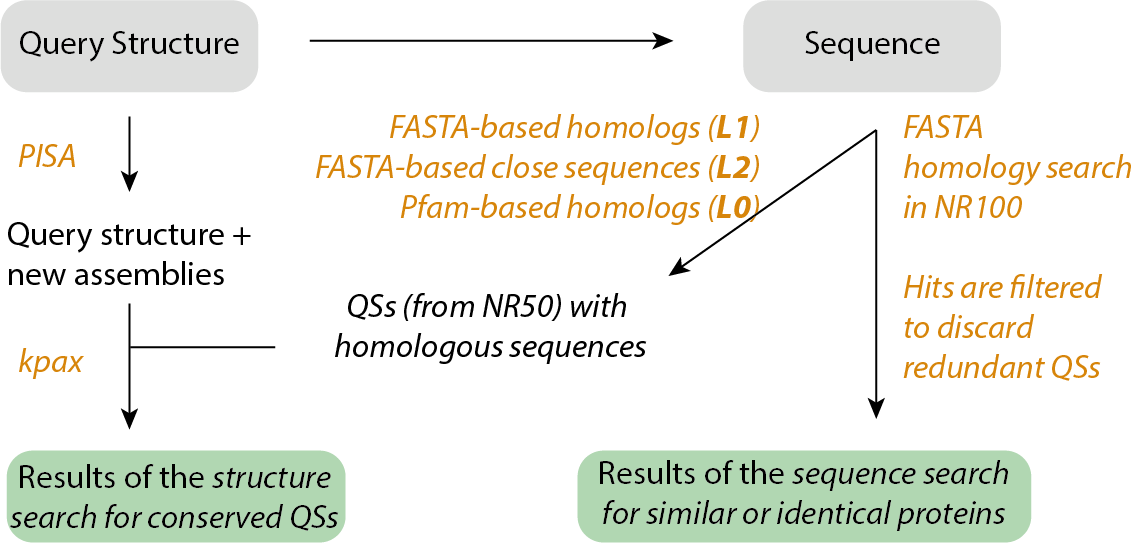
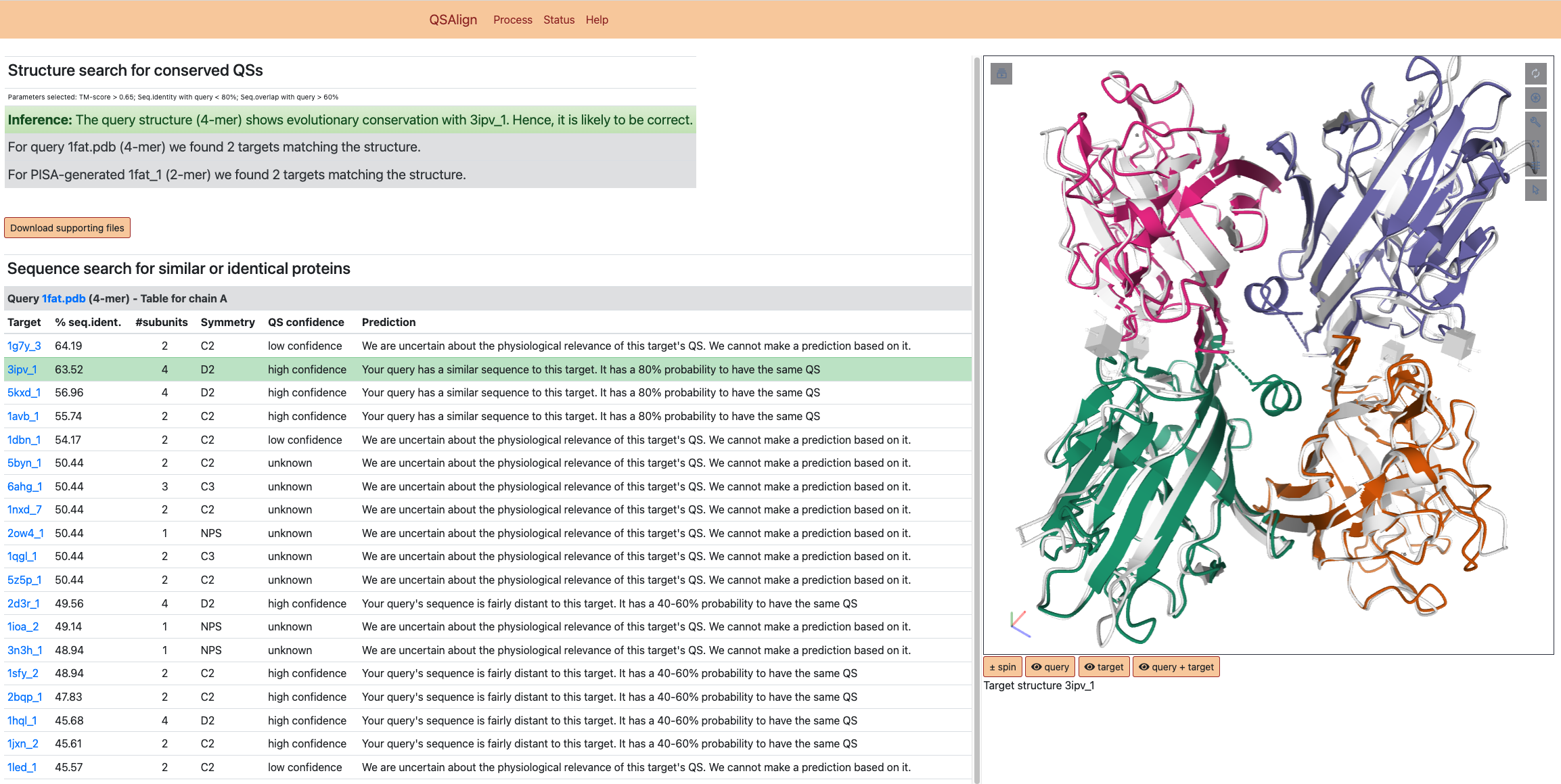
The user submits a query structure. Additional assemblies are identified using PISA. The resulting assemblies are each superposed with target QSs identified by sequence homology, based on Pfam domain similarity and sequence similarity. Ultimately, we display information on QS geometry conservation with the targets under the section " Structure search for conserved QSs". We also provide a table of representative QSs that share sequence homology with the query or that can even be identical to it. These are under the section Sequence search for similar or identical proteins.
4. How to interpret the results
The result page of QSalignWeb consists of two sections: This first one describes the prediction made based on the superposition with homologous QSs. The superposition of the two QSs based on which the prediction is made is shown on the right-hand side. Underneath, a second section consists of a table. There we display the result of the search of non-redundant QSs with a sequence similar (or even identical) to that of the query. In this list, the closest homolog with a high-confidence QS is highlighted in green and represents the structure we judge best for homology modeling.
5. Extrapolating QS by homology
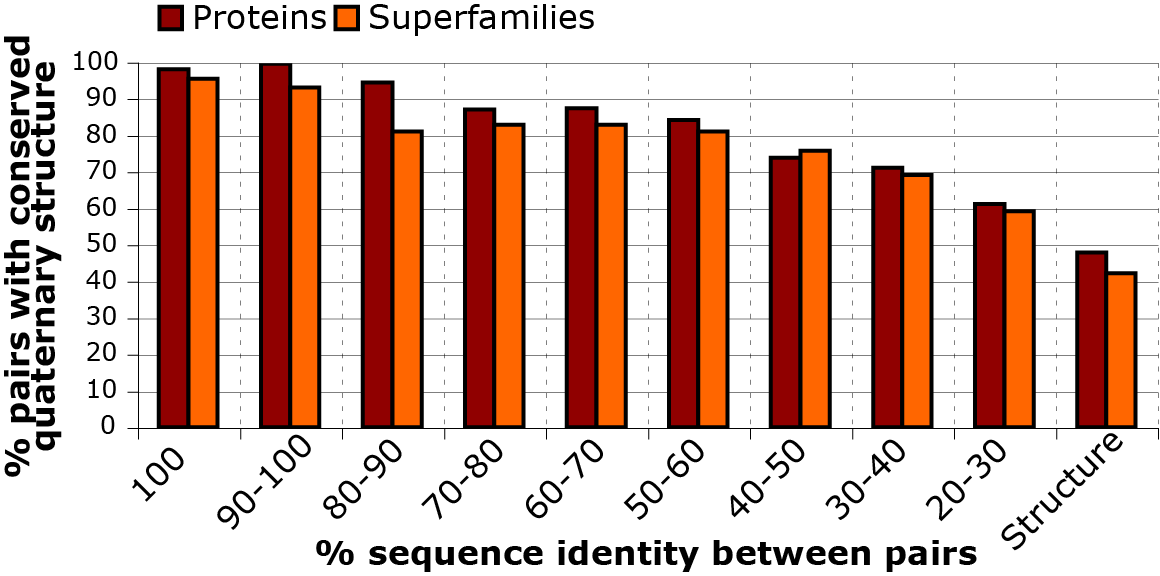
In the homolog results table, we predict the query sequence to adopt a particular QS based on the level of sequence identity. This prediction is based on the data below, which was published in Levy et al. 2008.