Outline

Most proteins act in concert with other proteins, forming permanent or transient
complexes. Understanding these interactions at an atomic level is only possible
through analysis of protein structures. Over the past few years, there have been
efforts to infer the quaternary structures of X-ray crystal structures (Henrick and
Thornton 1998; Valdar and Thornton 2001; Ponstingl, Kabir et al. 2003; Bahadur,
Chakrabarti et al. 2004), which support the prediction of the Biological Unit to the
structure in PDB.
Based on these predictions, we present a visualization and comparison strategy, and construct a hierarchical classification of complexes to integrate and organise the structures. Our strategy is organized in two main steps that we illustrate below:
We also discuss about the symmetry types that are found in protein complexes, and
brief explanations can be found here
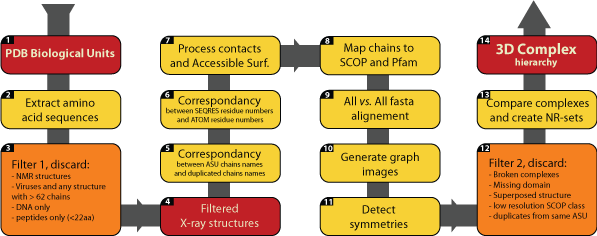
Flowchart highlighting the main steps involved in builing 3D Complex. The steps 10, 13 and 14, which are critical are discussed in more detail below Main steps involved in building 3D Complex
Representing Protein Complexes as Graphs
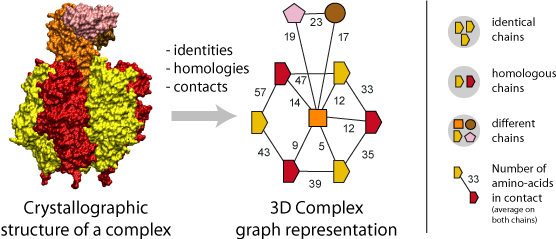
A fundamental step of our method is the translation of each protein complex into a graph. This involves processing the identities, homologies and contacts between the chains within each complex. Structural homology is detected using the N- to C-terminal order of SCOP superfamilies also called the domain architecture, and sequence identity is detected after a FASTA alignment with a 99% sequence identity threshold. Protein-protein interfaces are defined by a threshold of at least ten residues in contact, where the number of residues is the sum contributed by both chains. A residue-residue contact is counted if any pair of atomic groups is closer than the sum of their Van der Waals radii plus 0.5 � (Tsai, Taylor et al. 1999).
The graph itself thus provides the topology of the complex, i.e., the number polypeptides chains (nodes) together with their pattern of interfaces (edges), and an additional label associated to each graph carries the complex symmetry. Also, a label on each edge indicates the number of residues at the interface.
Importantly, the color and shapes of nodes are consistent within a graph but not among different graphs
Comparing Protein Complexes to construct a hierarchy
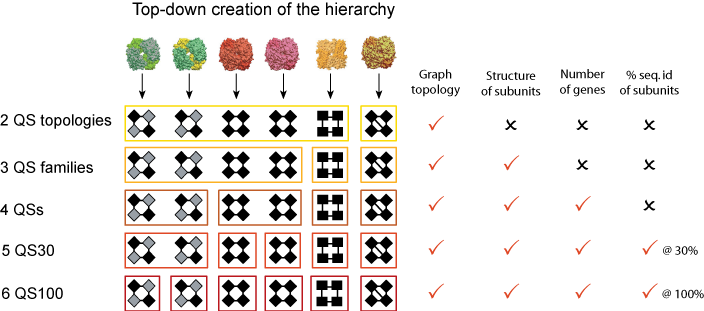
Once we have translated all protein complexes into their corresponding graph, we compare them with each other in order to build the hierarchy. From the graph representation, we have access to four types of information about each complex:
- The topology, represented by the number of nodes and their pattern of contacts,
- The structure of each constituent chain in the form of SCOP domain architecture,
- The amino acid sequence of each constituent chain, and thus the number of identical and homologous chains,
- The symmetry of the complex.
With these four types of information, we elaborate progressively stricter criteria that we use for comparing and grouping complexes, as illustrated in the figure above. First we group complexes according to their topology (e.g. their number of chains + pattern of contacts). Each of these groups is a QS Topology.
We then break down QS topologies into smaller groups called QS Families. In these, complexes have chains that are similar to each other structurally (e.g. same SCOP domain architecture). This means that two complexes in the same QS Family can be distantly homologous, sharing structural similarity but very little sequence identity. At the next level, we include the requirement that two matching complexes must have an identical number of genes. This is illustrated with hemoglobins in the figure above, where the hemoglobin α2β2 and the hemoglobin γ4 are in the same QS Family but in different QSs
From level four onwards, we require sequence homology between two matching chains, from 20% identity (4th level) to 100% (12th level). We call these groups QS20/30/40 etc. As the sequence similarity threshold gets stricter, the Quaternary Structure groups break down into smaller sub-groups.
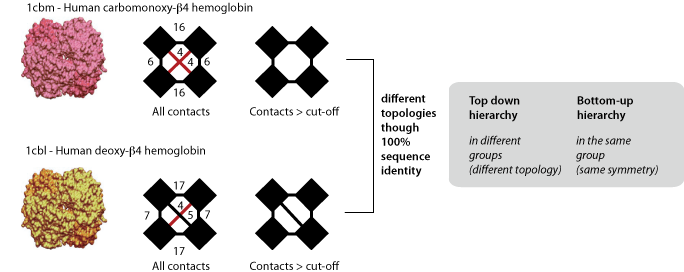
In top-down hierarchy, complexes with different topologies are never grouped together. This can be problematic in respect of the redundancy because closely related complexes can have different topologies, as illustrated with two human hemoglobin β4 homotetramers above. Therefore, in addition to the top-down hierarchy we provide a bottom-up hierarchy, where these two complexes are grouped together. The bottom-up hierarchy is identical to the top-down, except for homo-oligomers, which are grouped together on the basis of their symmetry instead of their topology. If you intend to carry out an analysis on homomers, we recommend the use of the bottom-up hierarchy.
The PDB Biological Unit
X-ray crystallography is the most common method used to gain information about protein structure. Structures obtained with this method correspond to an asymmetric-unit (ASU) i.e. the minimal unit from which the whole crystal can be reconstructed using symmetry operations. Importantly, the protein(s) contained in the ASU may not be in their native functional oligomeric state. For example, an ASU may contain a single copy of a protein that actually forms a homohexamer in vivo, or it might contain a dimer of a protein that exists as a monomer in vivo. This is why, in addition to the coordinates of the ASU, the Protein Data Bank makes available the coordinates of the Biological Unit, which is believed to contain the structure of the protein in its native state. For structures released up to 1998, the Biological Units are created automatically using the Protein Quaternary Structure server (PQS, (Henrick and Thornton 1998)). For structures from 1998 onwards, which are the majority of structures in PDB, the Biological Unit is manually curated and inferred based on the expertise of the authors of the structure as well as the PDB curators.
Symmetry of homomers
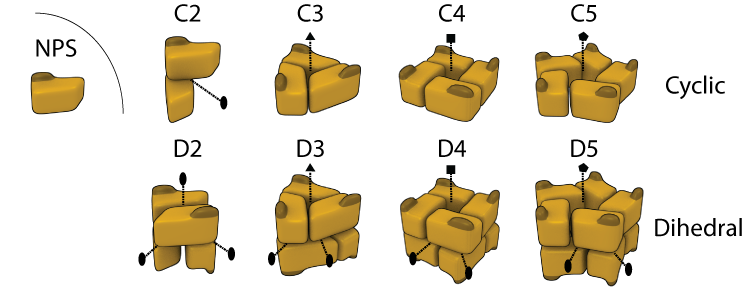
Homomers are very abundant in nature and also in 3D Complex, where they account for 90% of the complexes. They can be separated into two main classes of open or closed symmetry. The first class corresponds to open structures that would polymerize to infinity in the absence of limiting factors (not shown here). Such assemblies (e.g. tubulin and actin) are rare in 3D Complex (3%), most likely due to their innate dynamic character rendering them difficult to crystallize. By contrast closed symmetries are finite in space, and most homomers adopt either cyclic or dihedral symmetry (shown in Figure), with only a small fraction (1%) with cubic symmetry like virus' capsids (not shown). In 3D Complex we denote Cn a cyclic complex containing n subunits, and Dn as a dihedral complex containing 2*n subunits. In the figure, dotted lines with elipses, triangles, square and pentagons represent 2-, 3-, 4-, and 5- fold symmetry axes respectively.